Caso Clínico #53: torsades de pointes
Paciente de 59 años de edad.
Antecedentes: fibrilación auricular (bisoprolol y propafenona); insuficiencia aórtica grave, dislipemia y ex tabaquista 20 pack/year.
Cursando internación por neumonía por COVID-19 grave, con requerimientos de cánula de alto flujo por 24 horas y luego intubación orotraqueal al día +11 de síntomas. Inició tratamiento con dexametasona, tromboprofilaxis con enoxaparina (60 mg cada 12 hs), sedoanalgesia con midazolam, fentanilo y ketamina y bloqueo neuromuscular con rocuronio. Requirió una sesión de decúbito prono el día de conexión a ARM. Presentó durante la internación fibrilación auricular de alta respuesta ventricular, sin descompensación hemodinámica.
Evoluciona al día +6 de ventilación mecánica con episodio de taquicardia ventricular polimorfa no sostenida (12 latidos) evidenciada por monitoreo central, con reversión espontánea a ritmo de base (fibrilación auricular). Presenta un segundo episodio, sostenido (13 segundos) con hipotensión arterial leve (80/60 mmHg TA), que resuelve de forma espontánea nuevamente.


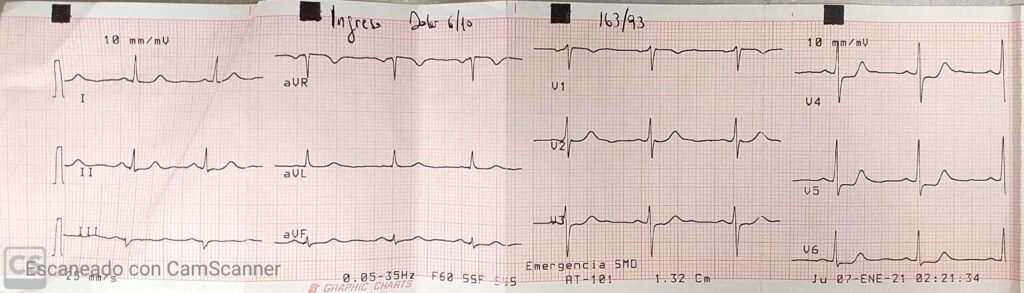
Se realiza ECG que evidencia ritmo de fibrilación auricular y la presencia de intervalo QT prolongado: 520 mseg (no evidenciado en ECG de ingreso).
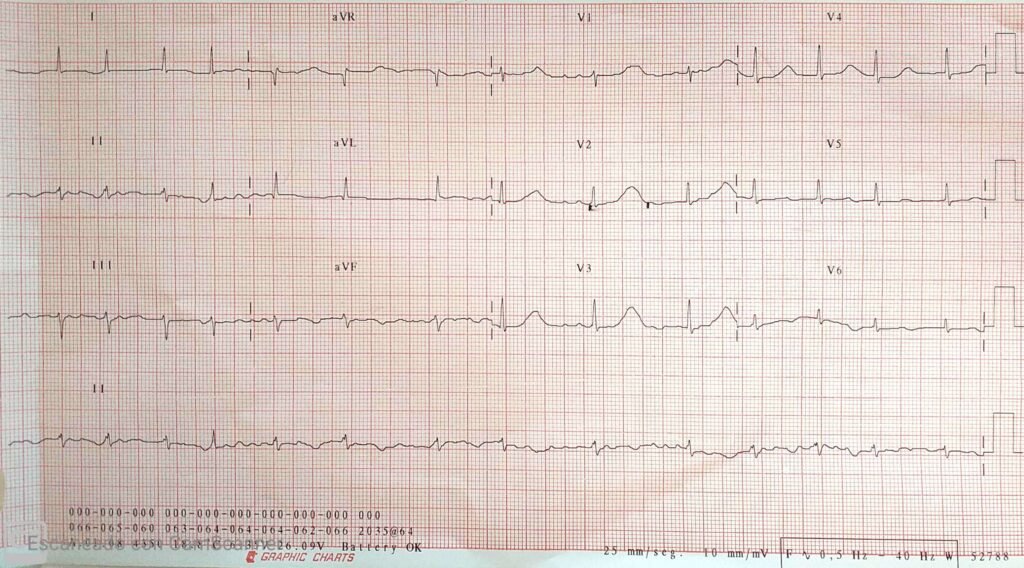
Nótese el intervalo QTc prolongado (520 ms)
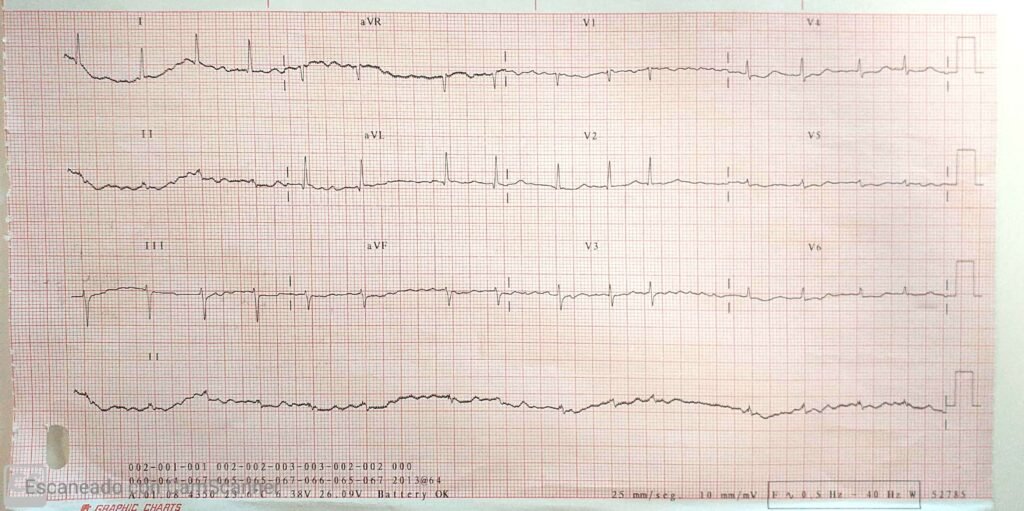
Nótese el intervalo QTc normal (430 ms)
Se interpreta evento como Torsades de Pointes (TdP) dado que se trataba de una taquicardia ventricular polimorfa en contexto de QT largo adquirido. Inicia tratamiento con sulfato de magnesio (2 gr de carga) sin complicaciones asociadas a la infusión.
Como posibles causas, se evalúan:
- Medicación predisponente: ese mismo día, el paciente había recibido, por primera vez en la
internación, olanzapina (dosis 5 mg), en plan de mejorar el weaning. Se hallaba en
tratamiento sedoanagésico con dexmedetomedina, pero no se encontraba bradicárdico con la
posibilidad de alargamiento del QT concomitante. - Alteraciones electrolíticas: se realiza laboratorio para descartar causas metabólicas: 4,1 mEq/l
de potasio; 2,1 mEq/l de magnesio; 8,4 mg/dl de calcio. - Alteraciones cardiovasculares: no presentaba pausas ni bradicardia sinusal (ritmo de
fibrilación auricular).
El paciente recurre con un tercer episodio de taquicardia ventricular que degenera en fibrilación
ventricular y consecuente paro cardiorrespiratorio, que tras maniobras de reanimación y una
cardioversión eléctrica revierte. Se coloca marcapasos transitorio para aumento de frecuencia cardíaca
y reducción de intervalo QT.
Los valores normales del QTc oscilan entre 350 y 450 milisegundos (ms) en los hombres y entre 360 y 460 ms en las mujeres. No es posible determinar el valor exacto de QTc que desencadenará una TdP pero el riesgo aumenta en la medida que se prolonga este intervalo [1].
La TdP es una forma específica de taquicardia ventricular polimorfa que se presenta en pacientes con intervalo QT largo (congénito o adquirido). Se caracteriza por complejos QRS rápidos e irregulares, que parecen torcerse alrededor del eje basal del electrocardiograma. La frecuencia cardíaca es de entre 150 y 300 latidos por minutos, con intervalos RR irregulares. Comienza de forma súbita y no es sostenida. Se detiene de forma espontánea o degenera en una fibrilación ventricular [2].
Este tipo de arritmia ventricular se produce secundario a causas congénitas o adquiridas [3]. Las congénitas se deben a alteraciones genéticas que codifican las proteínas de los canales transmembrana de sodio o de potasio. Esto genera un aumento en la carga positiva intracelular, que prolonga la repolarización ventricular y alarga el intervalo QT. Es uno de los causantes de muerte súbita infantil. Hay al menos 10 formas diferentes de sindrome de QT largo congénito. Las causas adquiridas son múltiples y variadas. Incluyen alteraciones hidroelectrolíticas (hipopotasemia, hipomagnesemia, hipocalcemia), fármacos (antiarrítmicos, macrólidos, antihistamínicos, neurolépticos, antidepresivos tricicíclicos), bradicardia sinusal o pausas por bloqueo sinoauricular o auriculoventricular, cardiopatía estructural (cardiopatía isquémica, hipertrofia ventricular), insuficiencia hepática o renal, cardioversión por fibrilación auricular y accidente cerebrovascular agudo.
El tratamiento consiste en la cardioversión en caso de que haya compromiso hemodinámico. Se deben retirar todos los fármacos que puedan alargar el QT y corregir posibles causas desencadenantes (alteraciones hidroelectorlíticas). El sulfato de magnesio es de primera elección y es independiente del nivel de magnesio sérico: un bolo de 2 gramos endovenoso en 2-3 minutos seguido o no de infusión continua. Si no hay respuesta, se puede pasar un segundo bolo (posibles efectos adversos: hipertensión y asistolia). Ya que el aumento de la frecuencia cardíaca acorta el intervalo QT, la infusión de isoproterenol endovenoso o la colocación de un marcapasos transitorio son opciones eficaces. Los pacientes con sindrome de QT largo congénito requieren tratamiento a largo plazo (betabloqueantes, marcapasos, cardiodesfibrilador implantable).
Como mensaje final, es fundamental realizar ECG al ingreso de los pacientes en las unidades críticas con medición del intervalo QT y su corrección por frecuencia cardíaca (60-110 según fórmula de Bazett y menor a 60 o mayor a 110 según Hodges) con una evaluación cuidadosa : 1) de los fármacos a implementar, debido a las posibles interacciones y alargamiento del QT como efecto adverso y, 2) del medio interno que frecuentemente está alterado en dichos pacientes.
Bibliografía
- Carreras Calvo F, Castellanos Rojas R, Perozo Panicello R, Ramírez Lana L. Síndrome
del QT largo y muerte súbita cardiovascular. Acta Med Colomb. 2015;19: 279–287. - Consenso de arritmias ventriculares. Revista Argentina de Cardiología, Vol 70, Suplemento 4 2002
https://www.sac.org.ar/wp-content/uploads/2014/08/Consenso-Arritmias-Ventriculares.pdf - Buller Viqueira E, Cabello Pulido J, Ibáñez Bulpe MJ. Torsade de pointes. Rev Clin Med
Fam. 2016;9: 63–67.