La vida del paciente trasplantado #10: síndrome hemofagocítico
Hombre de 71 años. ERCT secundaria a poliquistosis renal. Realizó hemodiálisis como terapia sustitutiva renal. Trasplante renal donante cadavérico. Serologías receptor: CMV (-), VEB (+), Chagas (-). Donante de 67 años, creatinina pre-ablación 1.8 mg/dl. Serologías donante: CMV (+), Chagas (+). Inducción con timoglobulina, metilprednisolona y micofenolato. Mantenimiento con belatacept, prednisona y micofenolato sódico. Profilaxis por 6 meses con valganciclovir por missmatch CMV. Creatinina habitual 1.7-2 mg/dl.
A los 6 meses del trasplante desarrolla proteinuria 1.7gr/24hs por lo que se realiza biopsia renal presentando rechazo celular agudo BANFF IA. Realiza tratamiento con pulsos de metilprednisolona. Anticuerpos Anti HLA I y II negativos.
A los 9 meses del trasplante presenta leucopenia y disfunción de injerto. PCR BK 16.000 copias/ml, log 4.2, PCR CMV <200 copias/ml. Se realiza biopsia de injerto: se evidencia Nefropatía por virus BK estadio II. Se rota inmunosupresión a prednisona y sirolimus y se agrega leflunomida. Creatinina habitual posterior a ese evento 2,5 mg/dl.
Luego cursó internación por infiltrados pulmonares y fiebre con diagnóstico por BAL de neumonitis por CMV. Recibió tratamiento con valganciclovir con cargas virales de CMV y BK indetectables. No realizó profilaxis secundaria para CMV por leucopenia.
A los dos años del trasplante, se interna por síndrome febril sin foco clínico. Laboratorio de ingreso: Hematocrito 16%, hemoglobina 5,6 g/dl, leucocitos 2.371/mm3, plaquetas 75.500/mm3, creatinina 3,27 mg/dl. LDH con valores entre 600-700 U/L, sin otros parámetros de hemólisis (bilirrubina normal y Coombs directa negativa) y Ferritina mayor a 5000 ng/ml. PCR para CMV detectable en sangre. Esplenomegalia de 177 mm por ecografía.
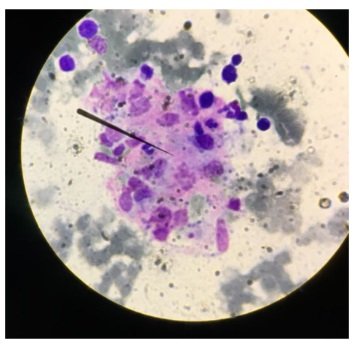
Se decidió realizar punción y aspiración de médula ósea. Se observó hemofagocitosis y proeritroblastos agrandados (ver figuras).

Inmunotipificación por citometría de flujo sin hallazgos de importancia. Se recibió informe de PCR para VEB y Parvovirus B19 detectable en material de médula ósea, PCR CMV y Chagas negativos en médula. Cultivos para gérmenes comunes, micológico, micobacterias y PCR para Tuberculosis negativos.
Se interpretó síndrome hemofagocítico desencadenado reactivo a infección viral por VEB y Parvovirus B19 y asociado a viremia por CMV. Se comenzó tratamiento antiviral con ganciclovir, se disminuyó la inmunosupresión quedando sólo con corticoides y se administró tratamiento con inmunoglobulina, dosis 35 g/día durante 5 días. Evolucionó favorablemente, afebril y con mejoría de las citopenias.
El síndrome hemofagocítico (o linfohistiocitosis hemofagocítica – HLH, según sus siglas en inglés) es una entidad hiperinflamatoria potencialmente mortal causada por una respuesta inmune altamente estimulada pero ineficaz. Su fisiopatología está pobremente entendida.
En la literatura, se describe tanto su forma primaria (múltiples alteraciones genéticas con herencia autosómica recesiva, que generan alteración en la capacidad citotóxica de linfocitos T y células NK con la consiguiente sobreproducción de interferón gamma, hiperactivación macrofágica y aumento de citoquinas que llevan al daño tisular masivo) como secundaria.
Las causas secundarias del síndrome hemofagocítico más frecuentes son:
– Neoplasias: linfomas, leucemias agudas
– Infecciones: virales (siendo el VEB la causa más frecuente), bacterianas diseminadas, fúngicas y parasitarias
– Enfermedades autoinmunes: síndrome de activación macrofágica (MAS)
– Enfermedades Metabólicas
Criterios diagnósticos HLH-2004
El diagnóstico de HLH puede establecerse si se cumplen uno de los dos criterios siguientes:
1. Diagnóstico molecular consistente con HLH
2. Criterios diagnósticos (deben cumplirse 5 de 8 de los siguientes):
– Fiebre
– Esplenomegalia
– Citopenias (que afecten 2-3 linajes celulares): Hb < 9 g/dl, Plaq < 100.000/m3, Neutrofilos < 1.000/m3
– Hipertrigliceridemia (>265 mg/dL) y/o hipofibrinogenemia (<150 mg/dL)
– Hiperferritininemia (>500 ng/mL)
– Hemofagocitosis en médula ósea, bazo o ganglios linfáticos
– Actividad baja/ausente de células NK
– Elevación CD25 soluble (receptor alfa de IL-2 soluble [sIL-2R]) dos desviaciones estándar por encima de las normas específicas de laboratorio ajustadas por edad
Existe poca bibliografía acerca del síndrome hemofagocítico en pacientes con trasplante renal. La mayoría de las publicaciones son casos reportados o serie de casos. En general, se coincide en el mal pronóstico de esta entidad, con una mortalidad que ronda el 50%.
Una serie de 17 pacientes trasplantados renales, publicada en el 2004, describió a las infecciones virales (CMV y VEB) como la causa más frecuente. El desarrollo del síndrome hemofagocítico ocurrió principalmente en contexto de aumento de la inmunosupresión, especialmente durante el período inicial después del trasplante, aunque también luego de tratamientos de rechazo. Esta relación es probablemente atribuible al mayor riesgo de complicaciones infecciosas o malignas asociadas con el uso de estos poderosos agentes inmunosupresores.
Si bien no hay consenso sobre la terapéutica óptima, un abordaje racional, planteado en un artículo publicado por Ponticelli y colaboradores en el 2009, consiste en maximizar el tratamiento antiviral (ej: Ganciclovir), disminuir la inmunosupresión (con excepción de los corticoides) y eventualmente administrar pulsos de corticoides (0,5 g por 3 días) o tratamiento con inmunoglobulina (0,4 g/kg/día durante 6-10 días). En casos refractarios se describe la plasmaféresis o leucaféresis.
El paciente descripto cumplía con 5 de los criterios diagnósticos descriptos anteriormente (fiebre, esplenomegalia, citopenias, hiperferritininemia y la presencia de hemofagocitosis en la punción de médula ósea). Tuvo una excelente evolución clínica con el tratamiento antiviral (ganciclovir, en contexto de viremia por CMV y la presencia de ParvovirusB19 y VEB En la médula ósea), la administración de inmunoglobulina y la disminución de la inmunosupresión, con mejoría de las series hematológicas y resolución del síndrome febril.
Bibliografía:
– Approach to Hemophagocytic Syndromes, Hematology Am Soc Hematol Educ Program, 2011, 178-83
– Hemophagocytic Syndrome in Renal Transplant Recipients: Report of 17 Cases and Review of Literature, Transplantation 77 (2), 238-43 2004 Jan 27
– Haemophagocytic Syndrome: A Life-Threatening Complication of Renal Transplantation, Nephrol Dial Transplant, 24 (9), 2623-7 Sep 2009