
Caso Clínico #19: autopsia virtual
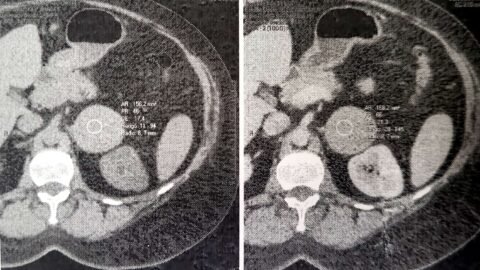
Mujer de 76 años, diagnóstico referido de Hipotiroidismo y Síndrome de Sjögren de larga data. Comienza su enfermedad actual hace aproximadamente 3 meses con síndrome de insuficiencia cardíaca. Se interna para estudio y se constata deterioro global de la función sistólica ventricular. Cinecoronariografía sin lesiones. Se realiza CardioRMN con gadolinio (ver imágenes): se informa miocardiopatía dilatada de causa no isquémico-necrótica con dilatación de cavidades izquierdas y FSVI deprimida gravemente; FSVD moderadamente deprimida, sin cavidades dilatadas; sin focos de fibrosis miocárdica; sin compromiso pericárdico.



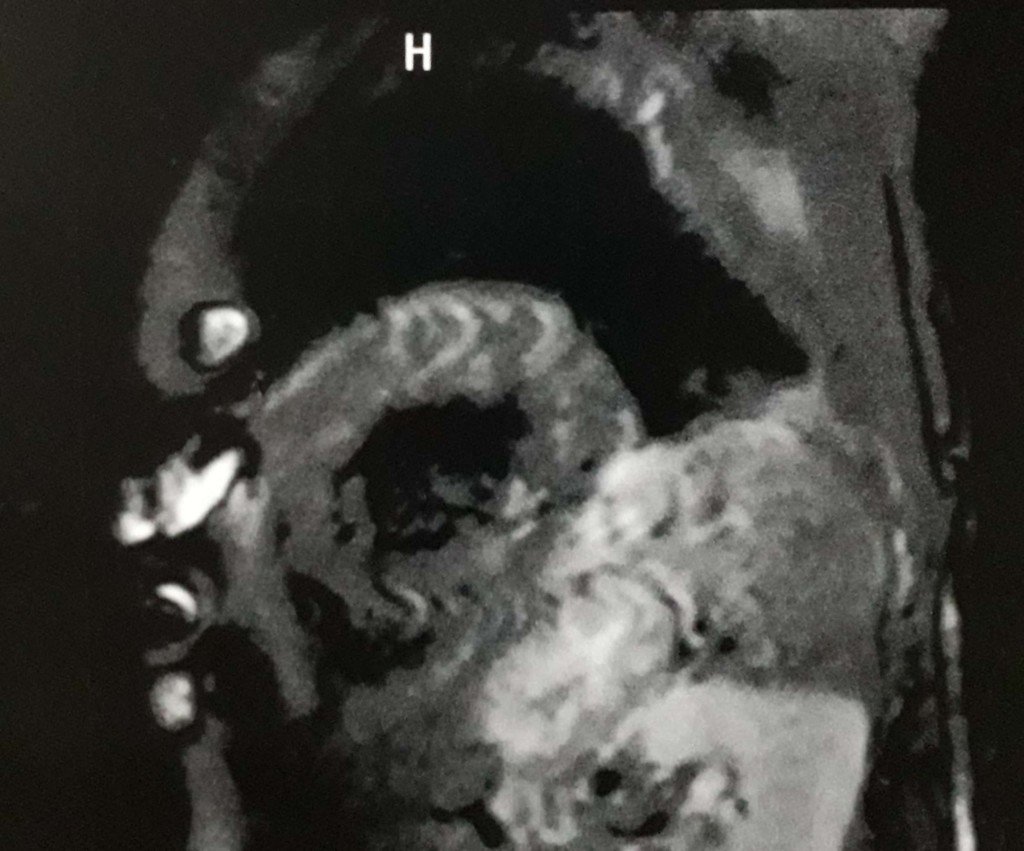
Presenta arritmia ventricular grave por lo cual requiere colocación de CDI. Además, presenta TVP femoral. Se optimiza tratamiento médico con sacubitrilo/valsartán, espironolactona y apixabán y se otorga alta.
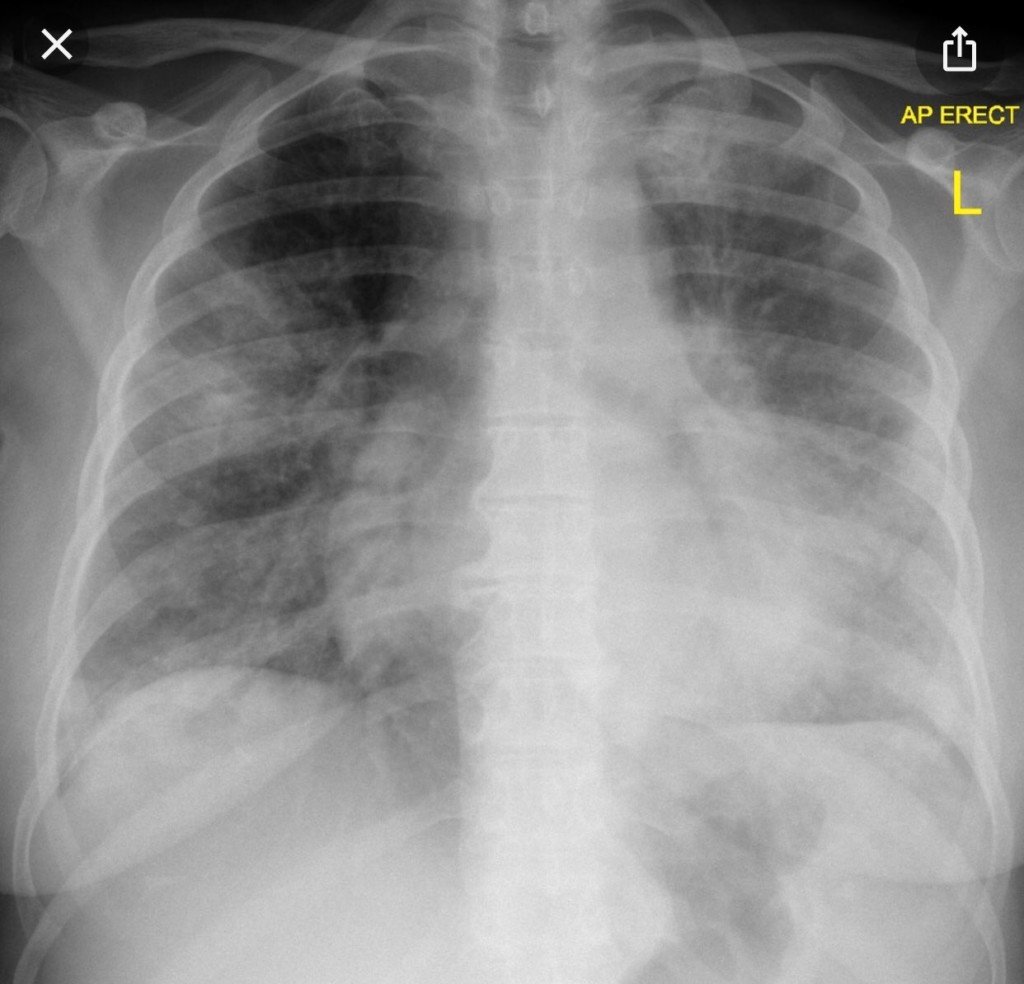
A los pocos meses se reinterna por encefalopatía en contexto de hiponatremia y sospecha de neumonía por infiltrados bibasales. Laboratorio con leucopenia, TSH 26 mUI/L, hipergammaglobulinemia policlonal de 2,9 g/dl. Recibe tratamiento con PTZ/oseltamivir con cultivos negativos. Evoluciona sin mejoría clínica. TC de encéfalo normal. PL con hiperproteinorraquia, resto de físico químico normal, cultivos negativos.
Presenta plaquetopenia y caída del Hto con parámetros de hemólisis: LDH >1000 U/L, haptoglobina ausente, Coombs directa positiva y esquistocitos en FSP. Por sospecha de Síndrome de Evans se inicia tratamiento con corticoides 1 mg/kg.
Evoluciona con injuria renal y persistencia de encefalopatía. Se plantea inicio de plasmaféresis por probable PTT. Laboratorio inmunológico: FAN + 1/640, antiDNA negativo, hipocomplementemia C3 y C4. Proteinuria 600 mg /24 hs. Requiere terapia sustitutiva renal por uremia. Mala evolución clínica a pesar del tratamiento instaurado, fallece a las 24 hs de iniciada la hemodiálisis.
El caso presentado no tuvo diagnóstico definitivo. Se invita a los miembros del foro a participar de la «autopsia virtual».
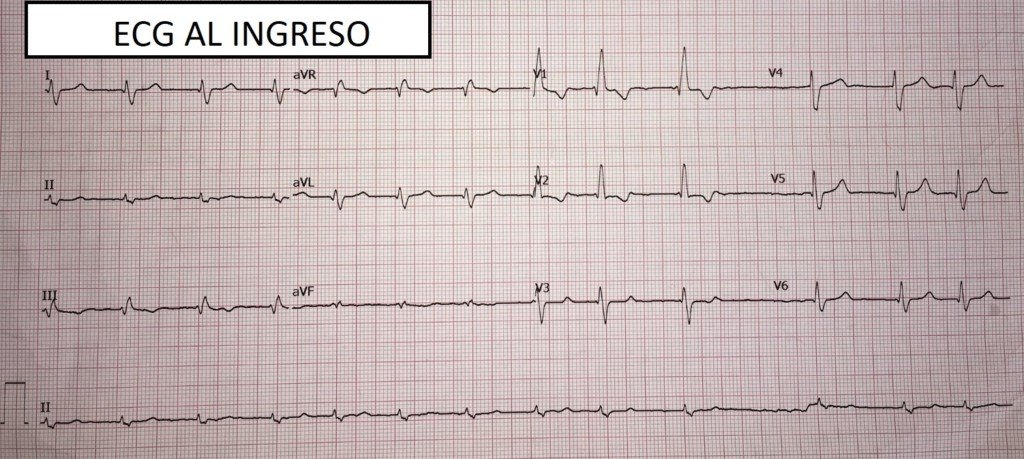




Muchas gracias por compartir este caso tan interesante. Me parece que hay mucho para discutir, y tengo algunas preguntas.
En primer lugar, se nos dice que la paciente tiene antecedentes de síndrome de Sjögren, cuán fuerte es ese diagnóstico? Se le hizo biopsia de glándulas salivares menores? Qué tipo de manifestaciones tuvo en el pasado? Glandulares solamente o extraglandulares? Estaba con tratamiento inmunomodulador o inmunosupresor previo a los sucesos que se relatan en el caso?
Su primer evento de importancia parece haber sido la insuficiencia asistólica a predominio izquierdo de causa desconocida. La hidroxicloroquina, que a veces se usa en el tratamiento del síndrome de Sjögren, puede causar miocardiopatía luego de décadas de uso. EL diagnóstico requiere biopsia endomiocárdica para ver las características específicas (vacuolas intracelulares y cuerpos curvilíneos en microscopio electrónica).
El siguiente evento es la internación con hiponatremia, encefalopatía e infiltrados bibasales. Sobre estos últimos, tenemos imágenes previas? Los infiltrados tenían apariencia intersticial? El SS está asociado a diferentes patrones de enfermedad pulmonar intersticial. Pudo ese haber sido el problema en los pulmones?
Por la descripción del caso no me queda claro la interpretación de la encefalopatía. Se consideró secundaria a la hiponatremia? Se hicieron bandas oligoclonales e índice de IgG en el LCR? Eso puede ser de utilidad para detectar inflamación en el LCR, aunque el conteo de blancos sea normales.
Luego, la pancytopenia de presumible origen autoinmune. Anemia hemolítica, con esquistocitos, haptoglobina indetectable, y trombocitopenia concuerdo señalan la posibilidad PTT. Se menciona la posibilidad de síndrome de Evans, los leucocitos también estaban bajos? Cuán bajos?
Acá hay que señalar que es bastante raro (pero no inaudito) que una persona con enfermedad autoimmune estable durante muchos años de repente tenga un curso agresivo en la vejez. Con los años el sistema inmune tiende más a la quiescencia, por lo que en este grupo etario, los problemas primariamente autoinmunes son raros. (Dicho esto, yo personalmente vi a una persona de 65 años con más de 30 años de lupus controlado sin tratamiento, tener un declive acelerado y morirse por la enfermedad en el espacio de unos pocos meses, y también vi un puñado de ancianos con lupus nuevo, así que… puede pasar).
Entonces tenemos que pensar si hay algún insulto subyacente que esté impulsando esta autoinmunidad. Particularmente, medicamentos, y neoplasias hematológicas. La paciente estuvo expuesta a medicamentos que se asocian con autoinmunidad en los últimos meses de su vida? (hidralazina, proacainamida, PTU, isoniazida, etc). Segundo, una de las complicaciones a largo plazo del SS es el desarrollo de linfomas (de células B generalmente). Un linfoma podría explicar bastante de lo que vemos. Había evidencia en las imágenes de linfadenopatías? Se hizo biopsia de médula?
Por último, la uremia. Se menciona preotinuria subnefrótica, había hematíes en la orina también? El SS puede causar nefritis intersticial. Había blancos en la orina?
El caso es muy interesante y me parece que la autopsia va a ser muy importante. Se va a realizar la autopsia?
Muchas gracias de nuevo
Ah, y otra cosa. Se chequearon antifosfolípidos? El síndrome antifosfolipídico catastrófico también podría explicar bastante de lo que vemos.
Hola. Dada la presencia de anemia hemolitica probablemente autoinmune y miocarditis (autoinmune?), se conoce algún otro dato del panel anti nuclear?
Hola a todos, se le atribuyó algún rol a esa TSH aumentada? Tal vez un hipotiroidismo severo (no si si tenía un fenotipo compatible) podría explicar algunos, no todos los signos encontrados en esta paciente. La cardiopatía con la consiguiente ICC, la predisposición a infecciones, la leucopenia, el deterioro del sensorio y la hiponatremia.
Saludos a todos, Joaquín.
Gracias a todos por participar. Intentaré ordenar un poco los pensamientos:
Retrospectivamente (y tarde), el equipo tratante planteó el diagnóstico de Lupus Eritematoso Sistémico.
Tomando los criterios clasificatorios SLICC, podemos ordenar las manifestaciones de la paciente de la siguiente manera:
– Proteinura >0,5/dl
– Anemia hemolitica (con Coombs directa positiva)
– Leucopenia (<4000/mm3)
- Trombocitopenia
- FAN positivo
- Hipocomplementemia
Si bien la miocarditis no se encuentra dentro de los criterios clasificatorios SLICC (sí la pericarditis), en este contexto podría interpretarse como dentro del mismo compromiso por el lupus.
Se postergó la realización de biopsia renal dado que la paciente se encontraba anticoagulada. Por otra parte, la autocrítica que realizó el equipo tratante fue el de no haber iniciado pulsos de corticoides ante la disfunción renal (sumada al compromiso hematológico y la "encefalopatía" nunca bien aclarada). Uno de los motivos que postergó el inicio de pulsos fue que la paciente ya se encontraba con corticoides, dosis 1 mg/kg, como tratamiento de lo que se había interpretado inicialmente como Síndrome de Evans.
Intentando responder algunos de los interrogantes planteados:
- Poco se conocía de su historia previa del Sjögren. No se contaba con biopsia de glándulas salivales y no se conocía ningún tratamiento específico. Tampoco estuvo expuesta a ningún medicamento que "gatille" autoinmunidad como planteó @juan-schmukler
– Los infiltrados pulmonares impresionaban de sobrecarga hídrica en zonas de decúbito (no se incluyeron aquí por cuestiones de espacio).
– En cuanto al panel antinuclear @santiago-vommaro: además del FAN +1/640 y el anti-DNA negativo, se solicitó AntiSm con resultado pendiente. El estudio del anticoagulante lúpico no se llegó a realizar.
– La TSH elevada se interpretó como tratamiento insuficiente con T4 dado la internación prolongada, problemas de absorción, etc @joaquin-castro
Lamentablemente no contaremos con autopsia ya que casi en ningún ámbito de medicina privada en la Argentina se cuenta con ello. Por eso nos pareció interesante intentar confeccionar una «autopsia virtual» del caso aquí en el foro.
Saludos y gracias nuevamente!
Hola, Martín. Gracias por la actualización. Comparto algunos de mis pensamientos posteriores.
Me cuesta ver este cuadro como de LES, por algunas de las razones que expliqué más arriba. Además, releyendo el caso, me cuesta encontrar evidencia para sostener la presunción de miocarditis. Tenía entendido (aunque puedo equivocarme) que la resonancia magnética cardíaca en casos de miocaditis muestra enhancement de contraste difuso reflejando la hiperemia por la inflamación difusa, lo que no se menciona en la descripción del caso.
La insuficiencia renal con proteinuria podría ser por lupus, pero en general cuando se ve un deterioro tan rápido de la función renal la lesión subyacente es glomerulonefritis, y no se menciona en el caso hematuria. Creo que el tema podría pasar más por un síndrome TMA, tipo antifosfolipídico catastrófico. Eso podría explicar la falla multiorgánica con pocas características inflamatorias y falta de respuesta a dosis altas de esteroides.
Por último, y para tranquilidad del equipo, hasta donde sé no existe evidencia que indique los pulsos de esteroides ofrecen beneficios adicionales a la dosis oral máxima de esteroides una vez que el tratamiento stá instaurado. por supuesto no existe evidencia de alta calidad que responda a esto directamente, pero en general se piensa en los pulsos como una “dosis de carga” más que una dosis supraefectiva de esteroides.
No sé, me cuesta pensar este caso como un lupus ultra rápido y letal, más a esta edad. Pero puedo estar equivocado.
Saludos.
El caso me parece sumamente interesante, como las reflexiones comentadas hasta aquí por todos. Me sumo a la discusión con algunas dudas y reflexiones propias.
No queda clara la etiología de la miocardiopatía dilatada que presenta la paciente y, como bien plantea Juan Schmuckler, la descripción de los hallazgos de la RMN no parecen corresponder a una miocarditis actual por la falta de realce ni a un compromiso previo por la ausencia de zonas de fibrosis. Tanto la miocarditis como las arritmias ventriculares son hallazgos muy poco frecuentes en el Sdme de Sjögren primario. Por otro lado, la disfunción tiroidea no parece ser causa suficiente para justificar este grado de deterioro de la función ventricular.
La presencia de citopenias es llamativa en esta paciente, ya que es una manifestación poco frecuente en el Sdme de Sjögren primario (10-20% para leucopenia, menos aún para trombocitopenia y menos aún para Sdme de Evans). Lo mismo ocurre con el compromiso de SNC, ya que el sdme confusional como manifestación de autoinmunidad es poco frecuente en el Sjögren y si bien podría explicarse por la hiponatremia, llama la atención la hiperproteinorraquia que presenta en el LCR.
En el Sdme de Sjögren, la aparición de nuevas citopenias podría hacer pensar en una progresión linfomatosa de la enfermedad, mas aun en presencia de factores de riesgo como la hipergammaglobulinemia y el complemento disminuido (otros factores de riesgo como la presencia de FR y crioglobulinas no han sido explicitadas en la descripción del caso). No obstante, en este caso el mecanismo de las citopenias no parece ser infiltrativo, por lo que desestimaría esta posibilidad.
La presencia de plaquetopenia y caída del Hto con parámetros de hemólisis con presencia de esquistocitos en FSP podría hacer pensar efectivamente en una microangiopatía trombótica (MAT), tipo púrpura trombótica trombocitopénica (PTT) adquirida. La presencia de esquistocitos aleja la posibilidad de hemolisis mediada por anticuerpos típico de una AHAI y hace sospechar en un mecanismo mecánico mas sugestivo de MAT. Como bien plantea Juan Schmuckler, este cuadro podría explicar la falla multiorgánica con compromiso renal, de SNC, citopenias y pocos marcadores de inflamación. El compromiso renal en MAT generalmente cursa con proteinuria leve e injuria renal aguda en los pacientes gravemente enfermos y el compromiso cardíaco cursa con insuficiencia cardíaca y arritmia en casos severos.
En términos de laboratorio inmunológico, si bien la presencia de FAN (+) es esperable (60-80% de los pacientes con Sjögren lo presentan), llama la atención la hipocomplementemia tanto para C3 como para C4 lo cual es poco frecuente en pacientes con Sjögren (si es mas frecuente la hipocomplementemia aislada para C4 en el contexto de crioglobulinemia por Sjögren).
Entre las causas de MAT, como bien se dijo previamente, habría que pensar en un sindrome antifosfolipídico catastrófico para lo cual el antecedente de la TVP podría sumar algunos puntos. No obstante, en la presencia de hipocomplementemia y FAN (+), podríamos pensar en una causa inmunomediada de MAT como la que puede verse en el caso del Lupus Eritematoso Sistémico con la aparición de anticuerpos anti inhibidores de la vía del complemento. Se nos dice que la paciente tenía diagnóstico de Sdme de Sjögren de larga data, pero me pregunto, ¿por qué no Lupus? El curso de la enfermedad parece demasiado agresivo para un Sdme de Sjögren y quizás mas coherente para un LES.
Si el cuadro se piensa en términos de una MAT, no resultaría tan raro que no haya respondido a esteroides VO (la AHAI/PTA probablemente si habrían respondido) y la paciente podría haberse beneficiado efectivamente del uso de plasmaféresis o inhibidores de complemento (Eculizumab).